|

牡丹,被誉为花中之王,是中国特有的世界著名花卉之一,也是传统中药材和重要的新兴油用资源作物,具有极高的经济价值、文化价值和社会价值。在基因组科学飞速发展的当今,仍然缺少高质量的染色体水平牡丹基因组。凤丹牡丹,即凤丹,又名杨山牡丹(Paeonia ostii),是中国最主要栽培牡丹(P. suffruticosa)的最重要祖先亲本之一,可赏、可食、可油用、可药用,也适用于多类高值产品开发;是当前中国生产和栽培面积最大的牡丹类型。与其它被子植物相比,牡丹(2n=10)具有千兆级的超级巨大的染色体(10-15 um)和非常大的基因组(>12Gb);由于过度开发利用(如野外采挖)以及其自身超大染色体和巨大基因组的影响,野生的凤丹植株已经不见踪迹。牡丹花具有大量离心发育的雄蕊,目前,在两千多栽培牡丹品种中,有上千个具有瓣化的雄蕊,雄蕊瓣化是牡丹花丰富多样的最重要途径之一,但这背后的机制依然是未知的。牡丹种子中含有大量的不饱和脂肪酸(>90%,如ALA),是人类自身不能够产生而又不可或缺的基本脂肪酸;尽管大量已有研究表明在陆地植物(包括牡丹)中脂肪酸的合成代谢路径十分保守,为什么牡丹中以ALA为代表的不饱和脂肪酸含量如此之高,至今依然不清楚。
2022年11月28日,国际著名期刊 Nature communication (IF = 17.694)在线发表了辰山植物园以 Genomic basis of the giga-chromosomes and giga-genome of tree peony Paeonia ostii (凤丹牡丹超大染色体及巨大基因组的遗传机制)为题的牡丹基因组学最新研究成果。该研究成功破解了凤丹牡丹高质量染色体水平的基因组遗传密码及其超大染色体形成与维持的潜在分子机制。这是迄今世界上已经测序的陆地植物中最大染色体(1.78-2.56Gb),也是已测序双子叶植物中最大基因组(12.28Gb)之一。此研究成果不仅将牡丹科学研究带入了真正的基因组时代,而且也开启了牡丹分子育种及优质特异基因鉴定和功能解析与产业利用的新纪元;是牡丹科学研究领域里程碑式的标志性成果,也是植物基因组学领域(巨大基因组和超大染色体研究)中最重要的突破性研究进展之一。

图1 凤丹牡丹超大染色体及巨大基因组特征图
1、历经十年艰辛探索、成功破解高复杂超大凤丹牡丹基因组遗传密码
牡丹基因组之复杂及破解难度之大,既在研究人员初始预料之中,但历经十年艰辛探索的周期之长却又极大的超出预料之外。这十年来,既是基因组科学及测序组装技术飞速发展的十年,也是凤丹基因组艰辛探索、逐步解析复杂谜团之旅。对凤丹牡丹基因组,我们采用了逐级梯度建库策略(150/300/500/800bp、2k/5/10k/20/40k文库)及二代高通量短片段测序技术(Illumia平台2.97Gb),结合三代长读长测序技术(PacBio平台 643.76Gb)和精准染色体构象捕获技术(HiC数据2.50Tb);在测序组装分析过程中,研究团队不断进行适用于牡丹复杂基因组特点的技术创新,并及时借鉴学习基因组学领域众多前沿技术,历经数百次/种软件测试、开发及应用分析和反复优化组装,曾先后形成过5个定型组装版本和3次完整(人工辅助)注释的拟发表版本。本文最终发表凤丹基因组成熟版本为12.28Gb(Contig N50=228Kb, Scaffold N50=2.43Mb),其中11.49Gb(约93.5%)成功组装到5条超大染色体(1.78-2.56Gb),这也是迄今人类已经测序陆地植物中最大的染色体。此版本中共注释基因73177条,高置信基因集59768条,有54451条锚定在5条不同染色体上。
2、逆转录转座子爆发式插入基因间区产生了牡丹超大染色体和巨大基因组
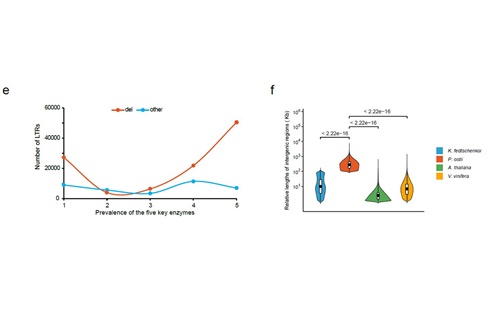
图2 凤丹牡丹中大量LTR爆发式插入基因组基因间区产生了超大基因组染色体
研究结果表明:在凤丹基因组中,约有330511条假基因和15238个基因家族,即据我们所知,牡丹基因组中假基因和基因家族的数量是迄今已经测序的植物中最多的物种。这些大量假基因的产生可能与LTR在基因组的大量扩增相关。与其它具有巨大基因组的单子叶植物大多经历了全基因组加倍事件相比,凤丹基因组似乎没有经历过其谱系特定的全基因组复制;而在短时间内(约 200 万年)其基因间区的逆转录转座子(以Del为代表的 LTR)爆炸性扩张是促成了其超大基因组和超大染色体的形成的可能机制。通过对16种代表性植物基因组中LTR的重新注释,本研究还提出了逆转录转座子Del家族相对于其它LTR亚家族在牡丹基因组中大量扩增及产生的可能机制,与其结构域的完整性(酶活性)相关。深入综合分析牡丹全基因组甲基化简化测序、组蛋白甲基化测序数据与LTR数据的结果表明,虽然牡丹拥有双子叶被子植物(迄今已测序)中最大的基因组和超级巨大的染色体,但其大部分功能基因依然可以正常的表达和转录,其原因就是大量的LTR是插入在远离功能区的基因间区。
3、凤丹牡丹千兆超大染色体的进化与形成机制
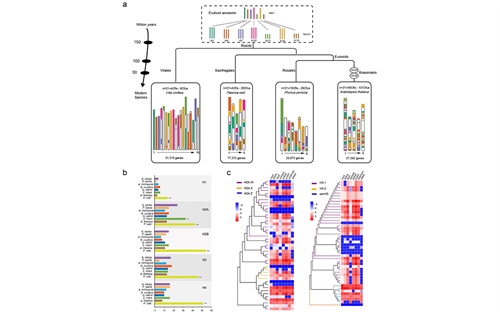
图3 凤丹染色体进化形成与维持机制
本研究采用被子植物12种代表性物种的系统进化分析结果,进一步明确了牡丹(芍药科)的系统位置处于核心双子叶植物基部的虎耳草目(Saxifragales),大约出现在109百万年前。4DTV进化分析表明以凤丹牡丹为代表的芍药科植物与葡萄等核心真双子叶植物可能共同经历了其祖先130百万年前的6倍化事件(γ)。同时,祖先染色推断与进化分析表明,牡丹基因组是通过祖先染色体(7条基数)至少经过4次断裂和20次融合等一系列复杂进化事件后才形成的当前牡丹类群中的5条染色体基数。研究表明:牡丹基因组中约有208个组蛋白编码基因(H1, H2A, H2B, H3 和H4),结合重新分析已发表的大量基因组数据,我们深入讨论了牡丹超大染色体的形成和维持所涉及的可能因素,本文中首次明确提出了牡丹这五种组蛋白编码基因的扩张(特别是H2A.W和H3.1)可能有助于维持其超级巨大的千兆染色体的初步科学猜想,且组蛋白数目与维持植物较大染色体相关的这一机制也可能存在普遍性,进一步的深入研究和更直接的证据仍在持续进行之中。
4、全基因组关联分析揭示了牡丹高不饱和脂肪酸之谜
同时,凤丹牡丹及其它芍药科植物还以其种子油而著称,其中富含不饱和脂肪酸,例如α-亚麻酸(ALA)。我们对 448 个种质进行了简化基因组测序和全基因组关联分析 (GWAS),并结合了种子时序发育转录组等技术,研究揭示了牡丹种子高效积累不饱和脂肪酸的重要机制,即其虽然与大多数陆地植物同样保守的脂肪酸生物合成途径,但是在通路中的每个关键节点至少有一个高表达基因在行使功能,进而保障了牡丹ALA等的大量积累;本研究还进一步鉴定了(包括SAD和FAD2等)多个候选油脂合成基因,可能在其种子高水平的ALA合成中发挥重要作用。本研究还系统解析了陆地植物中ALA合成关键环节(从亚油酸LA到α亚麻酸ALA)中FAD3和FAD7/8基因的进化历史,首次揭示了其在被子植物早期发生过一次基因复制事件及其重要意义。
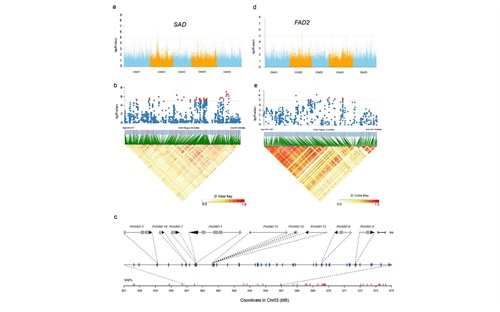
图4 GWAS揭示的牡丹高ALA合成相关的候选基因(SAD和FAD2)
5、栽培牡丹雄蕊瓣化的分子机制与人工驯化对牡丹花多样的贡献
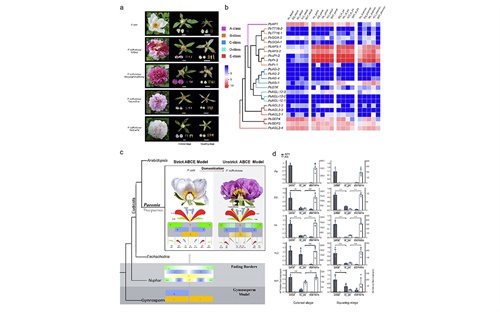
图5 牡丹花多样性及雄蕊瓣化的分子机制
凤丹牡丹(P. ostii)同时也是一种重要的观赏植物,是栽培牡丹(P. suffruticosa)的重要祖先亲本之一,本研究发现:花发育(器官身份决定基因)A类基因AP1的异位表达和C类基因AG在部分雄蕊中的表达减少可能有助于雄蕊瓣化的形成。同时,在栽培牡丹的长期栽培驯化过程中,决定花多样性的多个花器官发育基因明显受到人工选择压力。本研究进一步提出了牡丹花型发育的多样性模式假说,即在祖先亲本牡丹(P. ostii)的“花发育ABCE经典模型”(Strict ABCE Model)和栽培牡丹(P. suffruticosa)中的“不严格的花发育边界消退模型”(Unstrict ABCE Model), 结合选择分析结果,提出了牡丹花发育模式转变的进化驱动力就是栽培牡丹千百年来的人工驯化过程。这一研究发现,还进一步丰富了植物花发育研究的进化模式,即人工选择可能会打破前人研究表明的种子植物中花发育器官基因渠道化的方向性进化模式。该结果不但挖掘了栽培牡丹花多样性的多个候选基因,为进一步培育更优质和更高观赏价值牡丹提供了理论依据和基因资源。
总之,本研究成功破解了凤丹牡丹超大基因组/染色体的遗传密码,揭示了凤丹牡丹(P. ostii)不饱和脂肪酸生物合成相关的候选基因以及导致雄蕊瓣化形成的可能原因,其染色体水平的高质量基因组资源对于研究超大染色体/基因组进化和牡丹育种具有极其重要的科学及应用价值,是牡丹科学和植物基因组学研究领域的突破性、里程碑式的标志性成果之一。
上海辰山植物园袁军辉研究员(项目实际执行负责人,首席科学家)为本文第一作者及主要通讯作者,中科院植物研究所洪德元研究员和中科院分子植物卓越中心陈晓亚研究员为论文共同通讯作者(全程参与指导),牡丹课题组长胡永红教授为主要通讯作者(项目负责人)。华大基因姜三杰博士和简建波博士为本文共同第一作者。辰山牡丹组已毕业研究生刘铭妤和李娟(与青岛农业大学合培)、林黎虹和张林娟(与上海师范大学合培)、张晓骁和于水燕(原博士后)、张颖、张晓等;及岳震、徐加豹、许春艳、静一、陈海新、傅涛、吴章艳、王崇志、黄良博、王洪琦等(华大基因)为本文署名作者。牡丹基因组项目历时十年艰难探索,不仅是辰山植物园牡丹团队集体努力的成果,也是关注此项目的国内外各研究机构相关专家学者、牡丹专家及爱好者共同努力的结果。特别感谢牡丹专家李嘉珏先生、康仲英先生、付正林先生和魏春梅高工等提供部分实验材料;感谢韩斌研究员及团队成员、朱健康研究员、杨继教授、马红教授、孔宏智研究员、何跃辉研究员、张启翔教授、张大明研究员、张蘅研究员、焦远年研究员、李霖锋研究员等专家提供建议或参与了项目部分讨论;并向所有对项目有过帮助的(未署名)人员表示感谢。本研究得到上海市科委项目、国家自然基金及上海绿化市容管理局辰山专项资助。
基因组原文链接:
https://www.nature.com/articles/s41467-022-35063-1
上海辰山植物园牡丹组学与种质资源创新研究组是当前中国牡丹研究的主要生力军之一,主要研究方向:牡丹基因组学及各类特色性状(花型花色、药用及油用等)的遗传解析与关键基因挖掘、引种驯化与栽培起源研究、传统与分子育种、系统进化发育与保护生物学研究等。这也是辰山牡丹研究组近2多个月来,在国际知名植物学期刊上连续发表的第4篇高水平牡丹学术论文。
附文2:
2022年11月3日,国际植物学期刊《Frontiers in Plant Science》(IF=6.627,中科院2区)在线发表了题为“PacBio full-length sequencing integrated with RNA-Seq reveals the molecular mechanism of waterlogging and its recovery in Paeonia ostii”的研究论文。文章通过全长转录组测序和二代转录组测序相结合的技术,初步探究了凤丹响应淹水胁迫逆境及其淹水后恢复的分子机制,揭示凤丹以低氧静止综合征的策略来应对淹水胁迫的分子机制。
全文链接:
https://doi.org/10.3389/fpls.2022.1030584
附文3:
2022年10月11日,国际著名植物学期刊《Plant Biotechnology Journal》(IF=13.263,中科院1区)在线发表了牡丹组学在芍药苷生物合成途径解析研究中的一项突破研究进展,题目为“PoDPBT, a BAHD acyltransferase, catalyzes the benzoylation in paeoniflorin biosynthesis in Paeonia ostii”,成功克隆氧位苯甲酰基转移酶(基因)PoDPBT。
全文链接:
https://doi.org/10.1111/pbi.13947
附文4:2022年9月2于国际植物学期刊《Frontiers in Plant Science》(IF=6.627,中科院2区)发表了题为“Paeoniflorin in Paeoniaceae: Distribution, influencing factors, and biosynthesis”的综述论文。文章对芍药苷生物合成的后修饰途径进行了科学预测,并全面总结了芍药苷在芍药科植物中的分布及影响因素,并对芍药苷生物合成途径解析工作进行了展望。
全文链接:
https://doi.org/10.3389/fpls.2022.980854
诚邀有志于牡丹芍药研究的各类硕士、博士研究生及毕业生及出站博士后(或入站)到辰山学习、访问、联合培养或工作(联系人袁老师,yuanjunhui@csnbgsh.cn,长期有效)。
|

